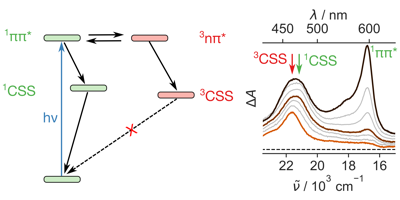
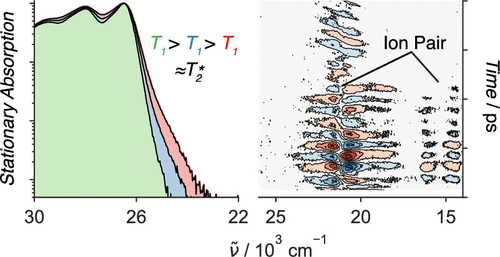
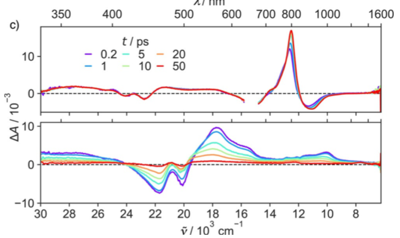
-
The excited-state dynamics of the radical anions of cyanoanthracenes

J.S. Beckwith, A. Aster and E. Vauthey
Physical Chemistry Chemical Physics, 24 (2022), p568-577


DOI:10.1039/D1CP04014F | unige:157670 | Abstract | Article HTML | Article PDF | Supporting Info
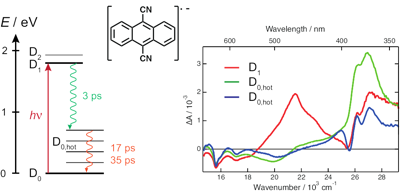
The radical anion of 9,10-dicyanoanthracene (DCA) has been suggested to be a promising chromophore for photoredox chemistry, due to its nanosecond excited-state lifetime determined from indirect measurements. Here, we investigate the excited-state dynamics of the radical anion of three cyanoanthracenes, including DCAÃâ¢Ã¢Ëâ, produced by photoinduced electron transfer in liquid using both pumpââ¬âprobe and pumpââ¬âpump probe transient electronic absorption spectroscopy. All three excited radical ions are characterised by a 3ââ¬â5 ps lifetime, due to efficient non-radiative deactivation to the ground state. The decay pathway most probably involves D1/D0 conical intersection(s), whose presence is favoured by the enhanced flexibility of the radical anions relative to their neutral counterparts. The origin of the discrepancy with the nanosecond lifetime of DCAÃâ¢Ã¢Ëâ* reported previously is discussed. These very short lifetimes limit, but do not preclude, photochemical applications of the cyanoanthracene anions.